Pediatric Dependency Map
Dharia et al demonstrate a first generation pediatric cancer dependency map that suggests that pediatric cancers have vulnerabilities that are distinct from those in adult cancers.
Our laboratory focuses on the integration of “omic” approaches for the identification of new protein targets and small-molecule modulators of cancers affecting children with an eye toward clinical translation.
We have focused our attention on pediatric malignancies notable for aberrancies of differentiation and/or oncogenic activation of transcription factors in an otherwise simple genomic background: the acute leukemias, Ewing sarcoma, and neuroblastoma. Our team is highly collaborative and draws on the expertise of cancer biologists, computational biologists, translational researchers, genomics scientists, and chemistry colleagues. Several major approaches used by our laboratory and exemplary discoveries emerging from our work are outlined below:
In order to overcome the challenges of traditional target- and phenotype-based screening, we previously developed a chemical genomic screening approach, Gene Expression-based High-throughput Screening (GE-HTS), in which gene expression signatures serve as surrogates for different biological states. Gene expression signatures of the biological states of interest are defined using genome-wide expression profiling; a high-throughput, low-cost signature assay is developed; and then a small-molecule library is screened for compounds that induce the desired signature switch. We have applied GE-HTS to two primary avenues of investigation in childhood malignancy: the discovery of modulators of biological state switches, such as differentiation, and the modulation of aberrant transcription factors. These GE-HTS screens, for example, led to the discovery of spleen tyrosine kinase (SYK) as a new target in acute myeloid leukemia (AML) and to the discovery of the SERCA channel inhibitor, thapsigargin, as an approach to inhibiting mutated NOTCH1 in T-cell acute lymphoblastic leukemia (T-ALL).
Our laboratory has collaboratively led efforts to characterize the genomic landscape of pediatric malignancies using massively parallel sequencing. For example, we were among the first groups to characterize the genomic landscape of Ewing sarcoma tumors. We determined that Ewing sarcoma tumors are some of the most genomically simple reported to date with a marked paucity of immediately targetable events in signal transduction pathways. However, STAG2 loss is present in more than 15% of Ewing sarcoma tumors and occurs by point mutation, rearrangement, and likely non-genetic mechanisms. STAG2 is a member of the cohesin complex, a protein complex critical for regulating sister chromatin exchange, chromatin architecture, and gene expression. Our studies, and that of others, have significant implications for clinical genomics efforts in this disease and also point to the importance of either targeting EWS/FLI or to identifying synthetic lethal relationships in the presence of EWS/FLI or mutations in STAG2.
We are currently leading a collaborative effort with the Broad Institute to characterize the key dependencies in a number of childhood cancers using functional, genome-scale screening approaches, including shRNA and CRISPR-Cas9 screening, integrated with high-throughput small-molecule library screening leveraging a collection of FDA-approved drugs and preclinical candidates. Our goal is to create a Pediatric Cancer Dependency Map that can be used by the larger pediatric cancer research community. Numerous exciting targets have emerged from this project, and the number of pediatric cancers included in our screen continues to expand.
Differentiation of acute myeloid leukemia (AML)
Despite dose intensified cytotoxic chemotherapy, including stem cell transplantation, only a minority of patients with AML will be cured of the disease. An alternative therapeutic approach is to induce differentiation of the leukemia cell. Differentiation therapy has been successful in treating acute promyelocytic leukemia (APL) by targeting with all-trans-retinoic acid (ATRA) and arsenic trioxide the fusion present in the majority of APLs, PML-RARα. Emerging data support a role not only for transcription factor-directed therapy, but also for kinase-directed therapy and the targeting of chromatin regulators, to induce AML differentiation. Using our GE-HTS approach, we discovered that the EGFR inhibitor gefitinib induces AML differentiation by an off-target mechanism. We next integrated proteomic and shRNA screening to identify the cytoplasmic kinase SYK as the relevant target. We have validated SYK inhibition as a new strategy for promoting AML differentiation, determined that the PI3K pathway is a downstream effector of SYK in AML and identified Integrin-beta 3 (ITGB3) as an upstream activator of SYK. Most recently, we determined that SYK is a critical regulator of FLT3, with FLT3 mutations a biomarker of response to SYK inhibition and SYK inhibitors and FLT3 inhibitors synergistic in AML. Two clinical trials testing gefitinib/erlotinib and two new trials testing SYK inhibitors in AML have resulted from these studies. We are now focused on understanding mechanisms of resistance to SYK inhibitors in AML and to identifying highly effective drug combinations.
A second AML differentiation project from our laboratory integrated the data from two independent small-molecule library screens and an shRNA screen. GSK-3α emerged as a target at the intersection of these three screens. GSK-3 is a constitutively active serine threonine kinase that inhibits the Wnt and hedgehog pathways while activating NFκB. Our data demonstrated that pan-inhibitors of GSK-3 induce AML differentiation. While these results point to GSK-3 as a target in leukemia, there are concerns in targeting GSK-3 in AML because pan-GSK-3 inhibition promotes β-catenin stabilization and thus would potentially promote self-renewal in AML progenitor cells. Moreover, other studies have shown that genetic loss of GSK-3β can promote myeloid malignancies. In our own studies and supported by others, we have shown that genetic loss of the alpha isoform in human AML does not lead to β-catenin stabilization whereas pan-GSK-3 inhibition does. Moreover, selective suppression of GSK-3α by multiple shRNAs induces differentiation, decreases AML cell growth, and attenuates colony formation in methylcellulose and AML progression in mouse models. Thus, a parsimonious approach to translating our findings to the clinic might be to develop a GSK-3 isoform-specific inhibitor, an effort that is ongoing.
Differentiation of neuroblastoma
A second pediatric malignancy notable for the integration of differentiation therapy (i.e., 13-cis-retinoic acid) into combination chemotherapy is neuroblastoma, the most common extracranial solid tumor in childhood. We extended the GE-HTS approach to the discovery of new drugs to differentiate neuroblastoma. Our first study identified the combination of HDAC inhibitors with 13-cis-retinoic acid as an approach to promote neuroblastoma differentiation while our most recent screens determined that differentiation could be induced with a selective HDAC 1/2 molecule avoiding the need of a pan-HDAC inhibitor. Moreover, HDAC2 scored as a top dependency in neuroblastoma compared to over 300 other cancer cell lines screened in a genome-scale CRISPR-Cas9 screen.
Highlights of our recent work.
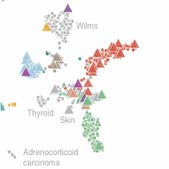
Dharia et al demonstrate a first generation pediatric cancer dependency map that suggests that pediatric cancers have vulnerabilities that are distinct from those in adult cancers.

Malone et al shows that selective modulation of the nuclear mRNA export protein NXT1 is a selective dependency in MYCN-amplified neuroblastomas.

Iniguez et al show that EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma.

Pikman et al demonstrate in a prospective clinical trial that systematic precision medicine enables matching of patient tumor mutations with targeted therapies.
